Ralph A. Wheeler
Computational Chemistry
Our research group is interested in developing better battery materials and optimizing enzymatic reactions. The central theme is understanding how non‐covalent interactions modulate chemical reactions as simple as ionic motion through solvents and as complex as methane oxidation to methanol by the enzyme methane monooxygenase. Students in my research group learn quantum mechanical, molecular dynamics, and/or QM/MM computational methods, as well as new ways to analyze their results. Former group members have held fellowships from the NSF, NIH, and National Research Council, as well as Goldwater Scholarships and Fulbright Fellowships. They have gone on to work in industry, government labs, teaching universities and research universities.
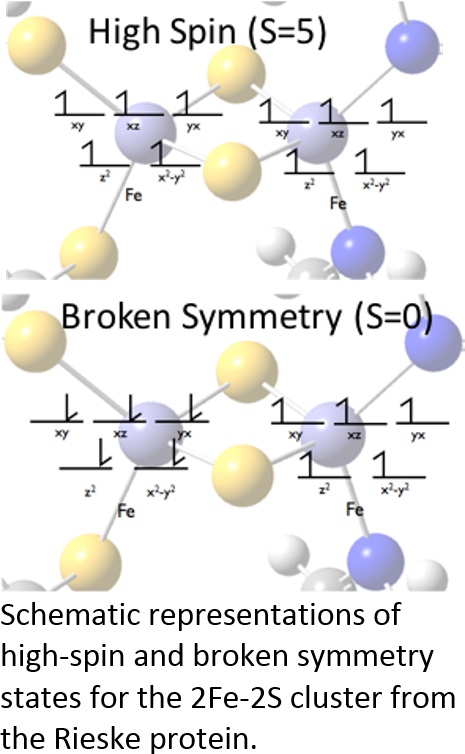 Our recent work to study enzymatic reactions has centered on implementing the extended broken symmetry (EBS) approximation with the quantum chemistry computer program Gaussian09. Using Gaussian's external optimizer feature, we wrote a program that weights energies, forces and force constants for the high spin and broken symmetry states to find the geometry of the ground singlet state. We tested the EBS approximation for 2Fe–2S clusters from ferredoxin, Rieske proteins and the more recently discovered mitoNEET proteins.1,2 Based on our calculations, we proposed that N‐15 isotopic labeling, combined with vibrational difference spectroscopy, may be used to distinguish experimentally the protonation states of histidine side chain ligands to the Rieske and mitoNEET clusters' iron atoms (this has important implications for proposed mechanisms of proton‐coupled electron transfer in these proteins). We are currently extending the method to model chemical reactions, including the environmentally and potentially industrially important reactions catalyzed by methane monooxygenase.
Our recent work to study enzymatic reactions has centered on implementing the extended broken symmetry (EBS) approximation with the quantum chemistry computer program Gaussian09. Using Gaussian's external optimizer feature, we wrote a program that weights energies, forces and force constants for the high spin and broken symmetry states to find the geometry of the ground singlet state. We tested the EBS approximation for 2Fe–2S clusters from ferredoxin, Rieske proteins and the more recently discovered mitoNEET proteins.1,2 Based on our calculations, we proposed that N‐15 isotopic labeling, combined with vibrational difference spectroscopy, may be used to distinguish experimentally the protonation states of histidine side chain ligands to the Rieske and mitoNEET clusters' iron atoms (this has important implications for proposed mechanisms of proton‐coupled electron transfer in these proteins). We are currently extending the method to model chemical reactions, including the environmentally and potentially industrially important reactions catalyzed by methane monooxygenase.
Recent massive recalls of consumer electronics due to faulty batteries could have been avoided by using safer, non‐flammable electrolytes. Ionic liquids are currently under study as new battery electrolytes due to their low flammability, low vapor pressure and the high ionic conductivity of dissolved lithium salts. Molecular dynamics simulations are currently underway to complement experimental work by the research groups of three collaborators. Our collaborators use (1) X‐ray diffraction to study long‐range ordering, (2) vibrational spectroscopy to study ionic aggregation and (3) measurements of transport properties (including viscosity, diffusion and ionic conductivity) to optimize properties of the ionic liquids in batteries. Analysis of our simulations currently focuses on understanding aggregation in and ordering of ionic liquids. Immediate extensions will include new ways to understand ionic conductivity, diffusion, and viscosity, as well as relations between them. Then, we plan to suggest chemical modifications to optimize ionic conductivity.
 1Jagger, B.R.; Koval, Ashlyn M.; Wheeler, R.A.; “Distinguishing protonation states of histidine ligands to the oxidized Rieske iron–sulfur cluster through 15N vibrational frequency shifts”; ChemPhysChem 2016, 17, 216–220; http://doi.org/10.1002/cphc.201500838.
1Jagger, B.R.; Koval, Ashlyn M.; Wheeler, R.A.; “Distinguishing protonation states of histidine ligands to the oxidized Rieske iron–sulfur cluster through 15N vibrational frequency shifts”; ChemPhysChem 2016, 17, 216–220; http://doi.org/10.1002/cphc.201500838.
Selected to be featured on the cover as one of the best papers in this issue.
 2Koval, Ashlyn M.; Jagger, B.R.; Wheeler, R.A.; “Distinguishing the protonation state of the histidine ligand to the oxidized iron–sulfur cluster from the mitoNEET family of proteins“; ChemPhysChem 2017, 18, 39–41; http://doi.org/10.1002/cphc.201600957.
2Koval, Ashlyn M.; Jagger, B.R.; Wheeler, R.A.; “Distinguishing the protonation state of the histidine ligand to the oxidized iron–sulfur cluster from the mitoNEET family of proteins“; ChemPhysChem 2017, 18, 39–41; http://doi.org/10.1002/cphc.201600957.
Selected to be featured on the cover as one of the best papers in this issue.

Professor
La Tourette Hall 418
815‐753‐1181
rwheeler5@niu.edu
Educational Background
Postdoctoral Research Fellow, University of Houston, 1987–1989
Ph.D., Cornell University, 1988
Research Interests
Computer modeling of metalloenzyme structures, properties and reactions; simulating properties of electrolytes for battery applications; adapting signal and image processing algorithms to analyze molecular dynamics simulations.
Group Members
|
Sarah Frye |
Ashley Koval |
Travis Mackoy |
Reese Richardson |



